Parmi les nombreux syndromes génétiques prédisposant au cancer, les patients atteints d’anémie de Fanconi, de dyskératose congénitale, de xeroderma pigmentosum, du syndrome de Li Fraumeni, du syndrome de Blooms, d’ataxie télangiectasie et du syndrome de Cowden ont montré une susceptibilité accrue au cancer de la bouche en raison de l’instabilité génétique.
Il existe des preuves solides montrant une prédisposition au cancer chez les patients présentant une anémie de Fanconi. La dyskératose congénitale (DKC) (également appelée syndrome de Zinsser-Cole-Engman) est une maladie héréditaire rare avec une prédisposition à la leucoplasie orale de la langue qui pourrait se transformer en cancer au début de la vie.
Dans ce chapitre, nous décrivons la susceptibilité au cancer de la bouche et aux lésions buccales à potentiel malin dans deux syndromes cancéreux, à savoir l’anémie de Fanconi et la dyskératose congénitale. Les lésions buccales à potentiel malin associés à ces deux syndromes sont la maladie chronique du greffon contre l’hôte (cGVHD) (récemment ajoutée à la classification des lésions à potentiel malin en 2020), le lichen plan oral et la leucoplasie orale.
L'anémie de Fanconi
- L’anémie de Fanconi (AF) est un trouble multisystémique caractérisé par un spectre d’anomalies congénitales, une insuffisance progressive de la moelle osseuse et une pancytopénie, une forte susceptibilité à la leucémie myéloïde aiguë (LMA) et aux tumeurs solides.
- L’AF est présente dans le monde entier et touche 1 naissance sur 100 000 aux États-Unis. Elle est notamment plus fréquente chez les Africains (environ 1 sur 22 000), estimée à 1 sur 45 000 en Israël et la population gitane espagnole présenterait la plus forte prévalence.
- L’AF est un syndrome cliniquement hétérogène d’insuffisance de la moelle osseuse, d’anomalies congénitales qui peuvent affecter tous les organes.
- Les patients atteints d’AF peuvent souffrir de diverses malformations. Les anomalies congénitales comprennent des malformations du squelette (petite taille, microcéphalie ou pouce hypoplasique), des anomalies des organes (rénaux, ophtalmiques, auriculaires, cardiaques et génitaux) et des anomalies de la pigmentation de la peau, comme les taches café au lait (tableau 1).
- Jusqu’à présent, 23 gènes ont été impliqués dans l’AF causée par des mutations germinales dans un des multiples gènes de l’AF (FANCA à FANCV). Les mutations dans le gène FANCA sont les plus courantes et sont impliquées dans 60 à 65 % des cas signalés. Toutes les mutations dans les gènes de l’AF sont transmises de manière autosomique récessive, à l’exception de FANCB et RAD51, qui sont transmises respectivement de manière liée au chromosome X et de manière autosomique dominante.
- Les protéines de l’AF interagissent dans une voie cellulaire commune et participent à divers aspects de la réparation de l’ADN, notamment la réparation des liaisons transversales de l’ADN, la stabilisation génomique et la régulation des protéines en aval. Les défauts de l’une de ces protéines de l’AF entraînent une instabilité génomique, des mécanismes de réparation de l’ADN défectueux et un risque accru de cancer.
- Les patients atteints de FA présentant un dysfonctionnement hématopoïétique doivent généralement subir une greffe de moelle osseuse ou une greffe de cellules souches hématopoïétiques , qui est l’option thérapeutique privilégiée.
- L’une des complications les plus courantes après une greffe de cellules souches hématopoïétiques est le développement de la maladie chronique du greffon contre l’hôte (cGVHD), qui est également un facteur de risque pour le développement de cancers, en particulier le carcinome épidermoïde buccal. La cGVHD est désormais reconnue comme une lésion à potentiel malin
- Étant donné le large éventail d’anomalies cliniques, les patients atteints d’AF nécessitent une évaluation complète au moment du diagnostic.
- L’AF peut être difficle à différencier d’autres syndromes génétiques, ce qui complique son diagnostic. L’expressivité variable de l’AF rend le diagnostic précoce difficile dans certains cas.
- Une description clinique détaillée des altérations les plus fréquentes trouvées dans les différents organes est donnée par Moreno et al (2021).
- Le diagnostic de laboratoire est établi par la positivité du test de cassure chromosomique lors de l’exposition à des agents de réticulation de l’ADN.
- Les autres méthodes de diagnostic utilisées sont le western blotting, l’amplification par PCR multiplexe et le séquençage de nouvelle génération.
- Une fois le diagnostic confirmé, une surveillance à vie des patients atteints d’AF est nécessaire pour assurer un diagnostic précoce du cancer. Le conseil génétique est essentiel.
CARACTÉRISTIQUES CLINIQUES DE L’ANÉMIE DE FANCONI
(adapted from Nalepa and Clapp 2018)
- Malformations cranofaciales
- Retard de développement
- Petite taille
- Hypodontie, petites dents
- Saignement buccal
- Gencives gonflées
- L’association VACTERL
- Pouce ou radius anormal
- Ostéoporose
- Insuffisance médullaire progressive
- Malformations cardiaques
- Malformations génito-urinaires et gastro-intestinales
- Ectopie ou dysplasie rénale
- Dysfonction endocrienienne
- Diminution de la fertilité
- Environ un quart des patients atteints d’AF qui développent des tumeurs solides ne sont diagnostiqués comme tels qu’après la découverte de leur cancer.
- Par conséquent, l’AF doit être envisagée dans les tumeurs malignes à apparition précoce. L’apparition précoce du cancer de la bouche, telle que décrite dans plusieurs rapports de cas (cités ci-dessous), peut servir d’indication pour envisager le diagnostic de l’AF chez les jeunes patients qui développent un HNSCC en l’absence de facteurs de risque.
- L’AF peut entraîner une insuffisance de la moelle osseuse. La transplantation de cellules souches hématopoïétiques peut entraîner une cGVHD.
- L’incidence estimée est de 200 à 800 fois plus élevée chez les patients atteints d’AF que dans la population générale. Récemment, les chercheurs se sont à nouveau intéressés au développement des carcinome épidermoïde de la cavité buccale dans l’AF.
- Dans une étude rapportant des cas d’AF, l’âge médian des 12 patients qui ont développé des carcinomes buccaux après une greffe de moelle osseuse était de 21 ans, ce qui était significativement plus jeune que l’âge de 28 ans pour les patients qui n’avaient pas reçu de greffe de moelle osseuse (P
- Masserot et al. (2008) ont décrit – la plus grande série à ce jour de carcinomes épidermoïdes de la tête et du cou – 13 patients (8 dans la cavité buccale) présentant une anémie de Fanconi après une transplantation de cellules souches hématopoïétiques (HSCT). La survenue d’un carcinome épidermoïde de la langue chez une jeune personne qui ne fume pas devrait inciter à considérer l’AF comme la condition médicale sous-jacente et conduire à la réalisation d’un test de dépistage des cassures chromosomiques.
La Dyskeratose Congenitale
La dyskératose congénitale (DC) est une génodermatose héréditaire multisystème rare entraînant une réduction de la fonction de la télémérase, ce qui nuit à la stabilité chromosomique. Également connue sous le nom de syndrome de Zinsser-Cole-Engman, elle a été décrite pour la première fois en 1906. Les télomères forment les extrémités des chromosomes et la télémérase est une enzyme responsable de la synthèse des télomères. Une réduction ou une altération de la fonction de la télémérase entraîne une réduction du matériel génétique et l’activation subséquente des « voies de réponse aux lésions de l’ADN », ce qui entraîne la mort cellulaire.
Une insuffisance de la moelle osseuse se produit car celle-ci est dépendante de la fonction télomérique en raison de son fort renouvellement cellulaire. En outre, la DC se manifeste aussi fréquemment par des signes cutanéo-muqueux, une fibrose pulmonaire et/ou hépatique.
- La dyskératose congénitale est l’un des rares syndromes héréditaires d’insuffisance médullaire qui présente des modes de transmission liés au chromosome X, autosomiques dominants et autosomiques récessifs.
- La mutation du défaut génétique spécifique DKC1 associée à la DC liée au chromosome X, la forme la plus grave de DC, entraîne l’absence de dyskérine fonctionnelle, ce qui altère gravement la fonction de la télémérase.
- Un certain nombre de mutations génétiques ont été identifiées et donnent lieu à différentes manifestations cliniques, notamment RTEL1 et TINF2, qui présentent respectivement une hypoplasie cérébelleuse et une maladie rétinienne.
- Les mutations récessives de NOP10, CTC1, NHP2, PARN et WRAP53 sont plus rares et entraînent des manifestations cliniques différentes.
Épidemiologie
- La dyskératose congénitale est rare. Elle se manifeste cliniquement entre 5 et 12 ans, avec une prédominance masculine.
- Une incidence estimée à 1 sur 1000000 personnes a été rapportée.
Manifestations cliniques
- La présentation de la DC est généralement précoce, contrairement aux formes acquises d’anémie aplasique, et elle est la conséquence de mutations génétiques.
- En 1906, Zinsser a décrit pour la première fois la triade caractéristique de la DC, qui comprend les éléments suivants :
- Atrophie réticulaire et hyperpigmentation cutanée
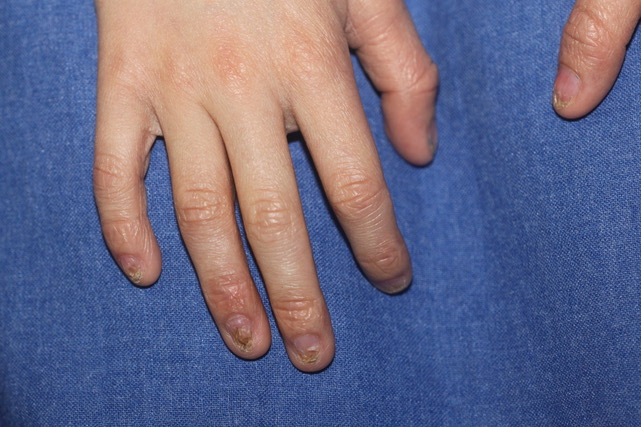
- Dystrophie des ongles (Figure 1)
- Leucoplasie orale
- Chez les patients atteints de DC en raison d’une instabilité chromosomique, il existe un risque accru de développer un cancer touchant divers organes.
- Il a été signalé que les enfants atteints de DC ont un risque 100 fois plus élevé de développer une leucémie myéloïde aiguë (LMA).
- Il existe également un risque accru de développer des carcinomes épidermoïdes de la tête et du cou, du col de l’utérus et de la région anogénitale.
- Des cancers de la peau, des voies aérodigestives et du pancréas ont également été signalés et la majorité de tous ces cancers surviennent au cours de la troisième décennie.
Manifestations buccales
- Un certain nombre de manifestations buccales ont été signalées chez les patients atteints de DC, notamment la leucoplasie orale, qui est la caractéristique la plus courante chez 80 % des patients.
- Les sites les plus fréquemment touchés sont la langue, la muqueuse buccale, le palais et les gencives.
- On peut également observer des modifications des tissus mous, notamment un érythème, en particulier au niveau de la muqueuse buccale et de la langue, une pigmentation brune et une dépapillation de la langue.
- En outre, une altération de la sensation décrite comme une brûlure de la langue a été signalée
Répercussion dentaire:
- L’inflammation gingivale et la maladie parodontale sont fréquemment observées chez les patients atteints de DC en raison de la combinaison d’une destruction précoce des tissus parodontaux résultant d’anomalies dans les structures dérivées ectodermiques, et d’une mauvaise réponse à cette destruction secondaire à la neutropénie.
- Les caries dentaires sont fréquemment identifiées chez les patients atteints de DC, une étude portant sur 73 patients ayant rapporté des » caries étendues » chez 13 d’entre eux, et ces résultats ont été confirmés par d’autres études.
- Les autres anomalies dentaires observées chez les patients atteints de DC sont l’hypodontie, des racines courtes et émoussées, l’émail aminci, le taurodontisme, la diminution du rapport couronne/racine, l’hypocalcification et la récession gingivale.
Transformation Maligne:
- Le taux de transformation maligne des leucoplasies orales a été documenté comme étant compris entre 0,13 et 34 %, en fonction de multiples facteurs de risque tels que l’âge, le sexe, l’homogénéité et la taille de la lésion.
- Dans la DC, le taux de transformlation de ces lésions en cancer est d’environ 30% entre 10 et 30 ans.
- De plus, en raison du raccourcissement précoce des télomères, on a signalé que les cancers se manifestent plus tôt chez des patients âgés d’à peine 5 ans. Par conséquent, une surveillance étroite est essentielle et nécessite souvent des biopsies.
Diagnostic différentiel
L’anémie aplasique peut être classée en deux grandes catégories : acquise et héréditaire. Les causes acquises comprennent l’idiopathie, qui est la cause la plus courante, les maladies médicamenteuses, virales, la grossesse et les maladies du tissu conjonctif. Les causes héréditaires comprennent l’anémie de Fanconi, le syndrome de Shwachman-Diamant et le syndrome GATA2. Bien que l’anémie de Fanconi reste le diagnostic différentiel le plus courant.
Les diagnostics différentiels des lésions blanches buccales impliquent des processus infectieux tels que la candidose et des causes non infectieuses, notamment le lichen plan buccal, les lésions lichénoïdes, le naevus blanc spongieux, la kératose frictionnelle et la maladie du greffon contre l’hôte GVHD.
Diagnostic
- es critères diagnostiques de la DC, tels que définis par Dokal 2011, stipulent qu’au moins 2 des 4 caractéristiques principales suivantes doivent être présentes (insuffisance médullaire, pigmentation anormale de la peau, dystrophie unguéale et leucoplasie) doivent être présentes, ainsi qu’un minimum d’au moins deux autres caractéristiques somatiques identifiées.Celles-ci sont présentées dans le tableau 1.
- Le diagnostic de DC peut être confirmé en effectuant des analyses sanguines, notamment une formule sanguine complète et une numération des réticulocytes, afin d’établir si une pancytopénie est présente.
- Une biopsie de la moelle osseuse peut révéler une moelle hypocellulaire.
- Les examens complémentaires comprennent des tests spécialisés tels que le séquençage génétique, la mesure de la longueur des télomères par cytométrie en flux, l’hybridation in situ en fluorescence (FISH) et la tomodensitométrie (CT) pour déterminer s’il y a une atteinte pulmonaire et/ou hépatique.
- En cas d’antécédents familiaux positifs, un diagnostic génétique prénatal par prélèvement de villosités choriales ou un diagnostic génétique préimplantatoire peut être proposé.
Prise en charge
- Une approche multidisciplinaire est nécessaire dans la prise en charge des patients atteints de la DC et différents professionnels de santé seront impliqués en fonction des organes concernés.
- Les hématologues auront un rôle central car l’insuffisance médullaire est une cause principale de morbidité et de mortalité précoces.
- La leucoplasie orale, en particulier chez les patients atteints de DC, a le potentiel de se transformer en cancer buccal et une surveillance attentive est donc nécessaire.
- Une approche globale sur la prévention et de la gestion des maladies dentaires par le biais de conseils est essentielle pour améliorer la qualité de vie des patients atteints de DC.
» Traitement non chirurgical
Traitement non chirurgical
La prise en charge non chirurgicale de la leucoplasie buccale avec la trétinoïne 0,1 % en solution topique a entraîné une amélioration partielle chez deux frères et sœurs âgés de 10 et 15 ans. Des agents chimiothérapeutiques tels que bleomycin and cyclophosphamide ont été signalés comme étant efficaces, mais les résultats ne se sont pas avérés cohérents pour améliorer la leucoplasie orale. Une autre étude a rapporté que l’étrétinate, un dérivé de la vitamine A, était efficace pour la leucoplasie orale chez les DC.
» View less
» Traitement chirurgical
Traitement chirurgical
Les chirurgiens oraux et maxillofaciaux peuvent décider d’intervenir chirurgicalement si des atypies ou un carcinome épidermoïde sont détectés. Les résections des tissus mous et durs de la bouche nécessitent souvent une reconstruction par prothèse, ce qui exige une planification détaillée avec un praticien en prothèse pour obtenir un résultat fonctionnel et esthétique satisfaisant.
| Caractéristiques cliniques/anomalies |
| Caractéristiques principales/communes |
| Pigmentation anormale de la peau |
| Dystrophie des ongles |
| Insuffisance médullaire |
| Leucoplasie |
| Autres caractéristiques somatiques reconnues |
| Epiphora |
| Difficultés d’apprentissage/retard de développement |
| Atteinte pulmonaire |
| Petite taille |
| Caries/pertes dentaires étendues |
| Sténose œsophagienne |
| Perte de cheveux prématurée/ cils grisâtres/secs |
| Hyperhidrose |
| Tumeur maligne |
| Retard de croissance intra-utérin |
| Maladie du foie/ulcération peptique/entéropathie |
| Ataxie/hypoplasie cérébelleuse |
| Hypogonadisme / testicules non descendus |
| Microcephalie |
| Rétrécissement/phimosis urétral |
| Ostéoporose/nécrose aseptique/scoliose |
| Surdité |
Tableau 1 : Caractéristiques cliniques utilisées dans les critères diagnostiques de la DC tels que décrits par Dokal (1).
» View less
Références et Lectures Complémentaires
Ankathil R, Mathew A, Joseph F, Nair MK (1996). Is oral cancer susceptibility inherited? Report of five oral cancer families. Eur J Cancer B Oral Oncol. 32(1):63–7. https://doi.org/10.1016/0964-1955(95)00055-0 PMID:8729621
Garavello W, Foschi R, Talamini R, La Vecchia C, Rossi M, Dal Maso L, et al. (2008). Family history and the risk of oral and pharyngeal cancer. Int J Cancer. 122(8):1827–31. https://doi.org/10.1002/ijc.23199 PMID:18076043
Warnakulasuriya S, Kujan O, Aguirre-Urizar JM, Bagan JV, González-Moles MÁ, Kerr AR, Lodi G, Mello FW, Monteiro L, Ogden GR, Sloan P, Johnson NW. Oral potentially malignant disorders: A consensus report from an international seminar on nomenclature and classification, convened by the WHO Collaborating Centre for Oral Cancer. Oral Dis. 2020 Oct 31. doi: 10.1111/odi.13704. Epub ahead of print. PMID: 33128420.
Moreno OM, Paredes AC, Suarez-Obando F, Rojas A. An update on Fanconi anaemia: Clinical, cytogenetic and molecular approaches (Review). Biomed Rep. 2021 Sep;15(3):74. doi: 10.3892/br.2021.1450. Epub 2021 Jul 15. PMID: 34405046; PMCID: PMC8329995.
Nalepa G, Clapp DW. Fanconi anaemia and cancer: an intricate relationship. Nat Rev Cancer. 2018 Mar;18(3):168-185. doi: 10.1038/nrc.2017.116. Epub 2018 Jan 29. PMID: 29376519.
Alter BP. Cancer in Fanconi anemia, 1927-2001. Cancer. 2003 Jan 15;97(2):425-40. doi: 10.1002/cncr.11046. PMID: 12518367.
Masserot C, Peffault de Latour R, Rocha V, Leblanc T, Rigolet A, Pascal F, Janin A, Soulier J, Gluckman E, Socié G. Head and neck squamous cell carcinoma in 13 patients with Fanconi anemia after hematopoietic stem cell transplantation. Cancer. 2008 Dec 15;113(12):3315-22. doi: 10.1002/cncr.23954. PMID: 18831513.
Millen FJ, Rainey MG, Hows JM, Burton PA, Irvine GH, Swirsky D. Oral squamous cell carcinoma after allogeneic bone marrow transplantation for Fanconi anaemia. Br J Haematol. 1997 Nov;99(2):410-4. doi: 10.1046/j.1365-2141.1997.3683184.x. PMID: 9375763.
Furquim CP, Pivovar A, Amenábar JM, Bonfim C, Torres-Pereira CC. Oral cancer in Fanconi anemia: Review of 121 cases. Crit Rev Oncol Hematol. 2018 May;125:35-40. doi: 10.1016/j.critrevonc.2018.02.013. Epub 2018 Mar 6. PMID: 29650274.
Kutler DI, Auerbach AD, Satagopan J, Giampietro PF, Batish SD, Huvos AG, Goberdhan A, Shah JP, Singh B. High incidence of head and neck squamous cell carcinoma in patients with Fanconi anemia. Arch Otolaryngol Head Neck Surg. 2003 Jan;129(1):106-12. doi: 10.1001/archotol.129.1.106. PMID: 12525204.
Amenábar JM, Torres-Pereira CC, Tang KD, Punyadeera C. Two enemies, one fight: An update of oral cancer in patients with Fanconi anemia. Cancer. 2019 Nov 15;125(22):3936-3946. doi: 10.1002/cncr.32435. Epub 2019 Aug 7. PMID: 31390058.
Grein Cavalcanti L, Fuentes Araujo RL, Bonfim C, Torres-Pereira CC. Oral manifestations compatible with chronic graft-versus host disease in patients with Fanconi anemia. Biol Blood Marrow Transplant. 2015a;21:275-280
Grein Cavalcanti L, Lyko KF, Araújo RL, Amenábar JM, Bonfim C, Torres-Pereira CC. Oral leukoplakia in patients with Fanconi anaemia without hematopoietic stem cell transplantation. Pediatr Blood Cancer. 2015b Jun;62(6):1024-6. doi: 10.1002/pbc.25417. Epub 2015 Feb 14. PMID: 25682760.
Bongiorno, M., Rivard, S., Hammer, D., & Kentosh, J. Malignant transformation of oral leukoplakia in a patient with dyskeratosis congenita. Oral Surgery, Oral Medicine, Oral Pathology and Oral Radiology 2017; 124(4), e239–e24
Handley, T. P., & Ogden, G. R. Dyskeratosis congenita: Oral hyperkeratosis in association with lichenoid reaction. Journal of Oral Pathology and Medicine, 2006; 35(8), 508–512. https://doi.org/10.1111/j.1600-0714.2006.00434.x
Ray JG, Swain N, Ghosh R, Richa, Mohanty SP. Dyskeratosis congenita with malignant transformation. BMJ Case Rep. 2011 Jan 11;2011:bcr0320102848. doi: 10.1136/bcr.03.2010.2848. PMID: 22715219; PMCID: PMC3029444
Ogden, G. R., Connor, E., & Chisholm, D. M. Dyskeratosis congenita: Report of a case and review of the literature. Oral Surgery, Oral Medicine, Oral Pathology, 1988; 65(5), 586–591. https://doi.org/10.1016/0030-4220(88)90142-9
McKay, G. S., Ogden, G. R., & Chisholm, D. M. (1991). Lingual hyperkeratosis in dyskeratosis congenita: Ultrastructural findings. Journal of Oral Pathology and Medicine, 20(4), 196–199. https://doi.org/10.1111/j.1600-0714.1991.tb00921.x
Ogden, G. R., Chisholm, D. M., Hopwood, D., & Lane, E. B. (1993). Evidence for field change in oral cancer based on cytokeratin expression. British Journal of Cancer, 67, 1324–1330. https://doi.org/10.1038/bjc.1993.245
Cannell H. Dyskeratosis congenita. Br J Oral Surg 1971;9: 8–10



