Of the many genetic cancer syndromes described patients with Fanconi anaemia, Dyskeratosis Congenita, xeroderma pigmentosum, Li Fraumeni syndrome, Blooms’s syndrome, ataxia telangiectasia, and Cowden syndrome have shown an increased susceptibility to oral cancer due to genetic instability.
Fanconi anaemia has the strongest evidence for a predisposition to cancer. Dyskeratosis Congenita (DKC) (also called Zinsser-Cole-Engman syndrome) is a rare hereditary condition with predisposition to oral leukoplakia of the tongue that could transform to cancer in early life.
In this chapter we describe susceptibility to oral cancer and oral potentially malignant disorders (OPMDs) in two cancer syndromes namely Fanconi anaemia and Dyskeratosis Congenita. OPMDs associated with these 2 syndromes are chronic graft vs host disease (cGVHD) (recently added to the OPMD classification in 2020), oral lichen planus and oral leukoplakia.
Fanconi Anaemia
- Fanconi anaemia (FA) is a multisystem disorder characterized by a spectrum of congenital abnormalities, progressive bone marrow failure and pancytopenia, high susceptibility to acute myeloid leukaemia (AML) and solid tumours.
- FA occurs worldwide and affects 1 in 100,000 births in the USA, and notably more common among Afrikaners (approximates 1 in 22,000), estimated at 1 in 45,000 in Israel and the Spanish Gypsy population is reported to having the highest prevalence.
- FA is a clinically a heterogenous syndrome of bone marrow failure (BMF), congenital abnormalities that may affect all organ systems.
- Patients with FA may suffer from a variety of malformations. Congenital anomalies include skeletal malformations (short stature, microcephaly, or hypoplastic thumb), organ abnormalities (renal, ophthalmic, ear, cardiac, and genital), abnormal skin pigmentation, like café au lait patches (Table 1).
- So far, 23 genes have been implicated in FA caused by germline mutations in one of multiple FA genes (FANCA to FANCV). Mutations in FANCA are the most common, involved in 60–65% of reported cases. All mutations in FA genes are inherited in an autosomal recessive manner, except for FANCB and RAD51, which are inherited in an X-linked manner and autosomal dominant manner, respectively.
- FA proteins interact in a common cellular pathway and participate in various aspects of DNA repair, particularly DNA crosslink repair, genomic stabilization, and regulation of downstream proteins. Defects in any of these FA proteins result in genomic instability, defective DNA repair mechanisms and an increased risk of cancer.
- FA patients with hematopoietic dysfunction usually require bone marrow transplantation (BMT) or hematopoietic stem cell transplantation (HSCT) which is the preferred treatment option.
- One of the most common complications after hematopoietic stem cell transplantation (HSCT) is the development of oral chronic graft-versus-host disease (cGVHD), which is also a risk factor for the development of cancer, particularly oral squamous cell carcinoma. cGVHD is now recognised as an oral potentially malignant disorder (OPMD)
- Given the wide range of clinical abnormalities, patients with FA require a comprehensive evaluation at diagnosis.
- FA may be grossly indistinguishable from other genetic syndromes, causing challenges to diagnosis. Variable expressiveness of FA makes early diagnosis difficult in certain cases.
- A detailed clinical description of most frequent alterations found in the different organs is given by Moreno et al (2021).
- Laboratory diagnosis is made by positivity in the chromosome breakage test upon exposure to DNA-crosslinking agents.
- Other diagnostic methods used include western blotting, multiplex ligation-dependent probe amplification and next-generation sequencing.
- Once the diagnosis is confirmed lifelong surveillance of patients with FA is required to ensure early diagnosis of cancer. Genetic counselling is essential.
CLINICAL FEATURES OF FANCONI ANAEMIA
(adapted from Nalepa and Clapp 2018)
- Craniofacial malformations
- Developmental delay
- Short stature
- Hypodontia, small teeth
- Oral bleeding
- Swollen gums
- VACTERL association
- Abnormal thumb or radius
- Osteoporosis
- Progressive BMF
- Cardiac defects
- Genitourinary and gastrointestinal malformations
- Renal ectopia or dysplasia
- Endocrine dysfunction
- Decreased fertility
- About a quarter of FA patients who develop solid tumours are diagnosed with FA only after discovery of their cancer.
- Therefore, FA should be considered in early-onset malignancies. Early-onset of oral cancer as described in several case reports (cited below) can serve as a pointer to consider the diagnosis of FA in young patients who develop HNSCC in the absence of risk factors.
- FA may lead to bone marrow failure, hematopoietic stem cell transplantation may lead to cGVHD.
- The estimated incidence is from 200-fold to 800-fold higher in patients with FA than in the general population. Recently, there has been renewed interest among researchers on development of OSCCs in FA.
- In a review of reported FA cases the median age of the 12 patients who developed oral carcinomas following a bone marrow transplant was 21 years which was significantly younger than the age of 28 for patients who had not received a bone marrow transplant (P< 0.02), suggesting a possible adverse effect of transplant ie cGVHD on the risk of developing an oral carcinoma (Alter,2003)
- Masserot et al., (2008) described – the largest series so far of head and neck squamous cell carcinomas – 13 patients (8 in the oral cavity) with Fanconi anemia after hematopoietic stem cell transplantation (HSCT). The occurrence of a squamous cell carcinoma of the tongue in a young person who does not smoke should prompt consideration of FA as the underlying medical condition and should lead to performance of a screening chromosome breakage test.
Dyskeratosis Congenita
Dyskeratosis congenita (DC) is a rare multisystem inherited genodermatosis leading to reduced telemerase function impairing chromosomal stability. Also known as Zinsser-Cole-Engman syndrome, it was first described in 1906 . Telemeres form the ends of chromosomes and telemerase is an enzyme responsible for the synthesis of telomeres. A reduction in or impaired function of telemerase leads to a reduction in genetic material and subsequent activation of ‘DNA damage response pathways’ resulting in cell death.
Bone marrow failure occurs as the bone marrow is dependent on telomere function due to its high cell turnover. In addition, DC also commonly manifests with mucocutaneous signs, pulmonary and/or hepatic fibrosis.
- Dyskeratosis congenita is one of a few inherited marrow failure syndromes which has inheritance patterns including X-linked, autosomal dominant and autosomal recessive.
- The specific genetic defect DKC1 mutation associated with X-linked DC, the most severe form of DC, leads to absence of functioning dyskerin, severely impairing telemerase function.
- A number of genetic mutations have been identified resulting in different clinical manifestations including RTEL1 and TINF2 which have cerebellar hypoplasia and retinal disease as prominent features, respectively.
- Recessive mutations in NOP10, CTC1, NHP2, PARN and WRAP53 are rarer and lead to differing clinical presentations.
Epidemiology
- Dyskeratosis congenita is rare, typically presenting clinically between 5-12 years and has a male predominance.
- An estimated incidence of 1 in 1000000 people has been reported.
Clinical Presentation
- The presentation of DC is typically early in life, in contrast to acquired forms of aplastic anaemia and are the consequence of the genetic mutations .
- Zinsser in 1906 first described the characteristic triad of DC which includes the following:
- Reticular atrophy and cutaneous hyperpigmentation
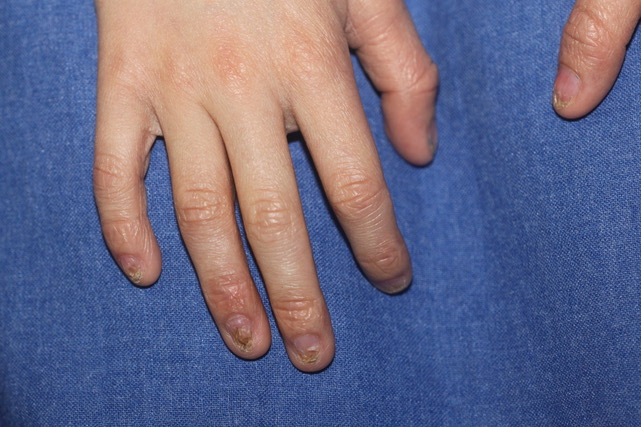
- Nail dystrophy (Figure 1)
- Oral leukoplakia
- In patients with DC as a result of chromosomal instability, there is an increased risk of developing cancer affecting various bodily systems.
- It has been reported that children with DC have a 100-fold increased risk of developing Acute Myeloid Leukaemia (AML).
- There is also an increased risk of developing squamous cell carcinomas of the head and neck, cervix and anogenital region.
- Cancers of the skin, aerodigestive tract and pancreas have also been reported and majority of all cancers occur in third decade of age.
Oral Manifestations
- A number of intraoral changes have been reported to present in patients with DC including oral leukoplakia, which is the most common feature found in up to 80% of patients .
- The most common sites involved are the tongue, buccal mucosa, palate and gingivae.
- There can also be soft tissue changes including erythema, particularly in the buccal mucosa and tongue, brown pigmentation, and depapillation of the tongue.
- In addition, altered sensation described as burning of the tongue has been reported
Dental Implications:
- Gingival inflammation and periodontal disease are frequently seen in patients with DC due to a combination of early destruction of periodontal tissues as a result of anomalies in ectodermal derived structures, and a poor response to this secondary to neutropenia.
- Dental caries is frequently identified in patients with DC with one study of 73 patients reporting ‘extensive caries’ in 13 patients and these findings have been supported elsewhere.
- Other dental anomalies seen in patients with DC include hypodontia, short blunted roots, thinned enamel, taurodontism, decreased crown/root ratio, hypocalcification and gingival recession.
Malignant Transformation:
- The rate of malignant transformation of oral leukoplakia has been documented to range between 0.13-34% depending on multiple risk factors such as age, sex, homogeneity and size of the lesion.
- In DC, the rate of progression of such lesions to cancer is approximately 30% between 10-30 years.
- Moreover, in DC due to early telomere shortening, cancers have been reported to present earlier in patients as young as 5 years. Therefore, close surveillance is essential and frequently require biopsies.
Differential diagnosis
Aplastic anaemia can be classified into two broad categories; acquired and inherited. The acquired causes include idiopathic, which is the most common cause, drug induced, viral, pregnancy and connective tissue diseases. Inherited causes include Fanconi anaemia, Shwachman- Diamond Syndrome and GATA2 syndrome. Though Fanconi anaemia remains the most common differential diagnosis.
Differential diagnoses oral white lesions involve infective processes such as candidiasis and non-infective causes including oral lichen planus, lichenoid lesions, white sponge naevus, frictional keratosis and graft-versus-host disease GVHD.
Diagnosis
- The diagnostic criteria for DC as outlined by Dokal 2011 states that at least 2 of 4 major features (BM failure, abnormal skin pigmentation, nail dystrophy and leukoplakia) must be present along with a minimum of at least two other identified somatic features. These are shown in the table 1.
- The diagnosis of DC can be confirmed by performing blood tests including full blood count (FBC) and reticulocyte count to establish if pancytopenia is present.
- A bone marrow biopsy may reveal a hypocellular marrow.
- Further investigations would include specialised tests such as genetic sequencing, measurement of telomere length by flow cytometry fluorescence in situ hybridisation (FISH) and computed tomography (CT) scans to determine if pulmonary and/or hepatic involvement.
- In the case of a positive family history, prenatal genetic diagnosis by chorionic villus sampling or preimplantation genetic diagnosis may be available.
Management
- Multidisciplinary (MDT) approach is required in the care of patient’s diagnosed with DC and the health care professionals involved will be governed by the bodily systems involved.
- Haematologists will have a focal role as bone marrow failure is a primary cause of early morbidity and mortality.
- Oral leukoplakia particularly in patients with DC has the potential to transform into oral cancer and therefore attentive surveillance is required.
- A holistic approach in preventing and managing dental disease through optimised preventative advice and interventions are essential in improving quality of life for patients with DC.
» Non-Surgical Management
Non-Surgical Management
Non-surgical management of oral leukoplakia with topical tretinoin 0.1% solution resulted in partial improvement in two siblings aged 10 and 15 years old. Chemotherapeutic agents such as bleomycin and cyclophosphamide have been reported to be effective but similar results have not shown to be consistent in improving oral leukoplakia. Another reported etretinate, a vitamin A derivative, was effective for oral leukoplakia in DC.
» View less
» Surgical Management
Surgical Management
Oral and maxillofacial surgeons may decide to surgically intervene if atypia or squamous cell carcinoma is detected. Resections of oral soft and hard tissue will often require reconstruction with prosthesis, therefore necessitating detailed planning with a prosthodontist to achieve a satisfactory functional and aesthetic result.
| Clinical features/abnormality |
| Major/common features |
| Abnormal skin pigmentation |
| Nail dystrophy |
| Bone marrow failure |
| Leucoplakia |
| Other recognised somatic features |
| Epiphora |
| Learning difficulties/developmental delay |
| Pulmonary disease |
| Short stature |
| Extensive dental caries/loss |
| Oesophageal stricture |
| Premature hair loss/greying/sparse eyelashes |
| Hyperhidrosis |
| Malignancy |
| Intrauterine growth retardation |
| Liver disease/peptic ulceration/enteropathy |
| Ataxia/cerebellar hypoplasia |
| Hypogonadism/undescended testes |
| Microcephaly |
| Urethral stricture/phimosis |
| Osteoporosis/aseptic necrosis/scoliosis |
| Deafness |
Table 1: Clinical features used in the diagnostic criteria of DC as described by Dokal (1).
» View less
References and Further Reading
Ankathil R, Mathew A, Joseph F, Nair MK (1996). Is oral cancer susceptibility inherited? Report of five oral cancer families. Eur J Cancer B Oral Oncol. 32(1):63–7. https://doi.org/10.1016/0964-1955(95)00055-0 PMID:8729621
Garavello W, Foschi R, Talamini R, La Vecchia C, Rossi M, Dal Maso L, et al. (2008). Family history and the risk of oral and pharyngeal cancer. Int J Cancer. 122(8):1827–31. https://doi.org/10.1002/ijc.23199 PMID:18076043
Warnakulasuriya S, Kujan O, Aguirre-Urizar JM, Bagan JV, González-Moles MÁ, Kerr AR, Lodi G, Mello FW, Monteiro L, Ogden GR, Sloan P, Johnson NW. Oral potentially malignant disorders: A consensus report from an international seminar on nomenclature and classification, convened by the WHO Collaborating Centre for Oral Cancer. Oral Dis. 2020 Oct 31. doi: 10.1111/odi.13704. Epub ahead of print. PMID: 33128420.
Moreno OM, Paredes AC, Suarez-Obando F, Rojas A. An update on Fanconi anaemia: Clinical, cytogenetic and molecular approaches (Review). Biomed Rep. 2021 Sep;15(3):74. doi: 10.3892/br.2021.1450. Epub 2021 Jul 15. PMID: 34405046; PMCID: PMC8329995.
Nalepa G, Clapp DW. Fanconi anaemia and cancer: an intricate relationship. Nat Rev Cancer. 2018 Mar;18(3):168-185. doi: 10.1038/nrc.2017.116. Epub 2018 Jan 29. PMID: 29376519.
Alter BP. Cancer in Fanconi anemia, 1927-2001. Cancer. 2003 Jan 15;97(2):425-40. doi: 10.1002/cncr.11046. PMID: 12518367.
Masserot C, Peffault de Latour R, Rocha V, Leblanc T, Rigolet A, Pascal F, Janin A, Soulier J, Gluckman E, Socié G. Head and neck squamous cell carcinoma in 13 patients with Fanconi anemia after hematopoietic stem cell transplantation. Cancer. 2008 Dec 15;113(12):3315-22. doi: 10.1002/cncr.23954. PMID: 18831513.
Millen FJ, Rainey MG, Hows JM, Burton PA, Irvine GH, Swirsky D. Oral squamous cell carcinoma after allogeneic bone marrow transplantation for Fanconi anaemia. Br J Haematol. 1997 Nov;99(2):410-4. doi: 10.1046/j.1365-2141.1997.3683184.x. PMID: 9375763.
Furquim CP, Pivovar A, Amenábar JM, Bonfim C, Torres-Pereira CC. Oral cancer in Fanconi anemia: Review of 121 cases. Crit Rev Oncol Hematol. 2018 May;125:35-40. doi: 10.1016/j.critrevonc.2018.02.013. Epub 2018 Mar 6. PMID: 29650274.
Kutler DI, Auerbach AD, Satagopan J, Giampietro PF, Batish SD, Huvos AG, Goberdhan A, Shah JP, Singh B. High incidence of head and neck squamous cell carcinoma in patients with Fanconi anemia. Arch Otolaryngol Head Neck Surg. 2003 Jan;129(1):106-12. doi: 10.1001/archotol.129.1.106. PMID: 12525204.
Amenábar JM, Torres-Pereira CC, Tang KD, Punyadeera C. Two enemies, one fight: An update of oral cancer in patients with Fanconi anemia. Cancer. 2019 Nov 15;125(22):3936-3946. doi: 10.1002/cncr.32435. Epub 2019 Aug 7. PMID: 31390058.
Grein Cavalcanti L, Fuentes Araujo RL, Bonfim C, Torres-Pereira CC. Oral manifestations compatible with chronic graft-versus host disease in patients with Fanconi anemia. Biol Blood Marrow Transplant. 2015a;21:275-280
Grein Cavalcanti L, Lyko KF, Araújo RL, Amenábar JM, Bonfim C, Torres-Pereira CC. Oral leukoplakia in patients with Fanconi anaemia without hematopoietic stem cell transplantation. Pediatr Blood Cancer. 2015b Jun;62(6):1024-6. doi: 10.1002/pbc.25417. Epub 2015 Feb 14. PMID: 25682760.
Bongiorno, M., Rivard, S., Hammer, D., & Kentosh, J. Malignant transformation of oral leukoplakia in a patient with dyskeratosis congenita. Oral Surgery, Oral Medicine, Oral Pathology and Oral Radiology 2017; 124(4), e239–e24
Handley, T. P., & Ogden, G. R. Dyskeratosis congenita: Oral hyperkeratosis in association with lichenoid reaction. Journal of Oral Pathology and Medicine, 2006; 35(8), 508–512. https://doi.org/10.1111/j.1600-0714.2006.00434.x
Ray JG, Swain N, Ghosh R, Richa, Mohanty SP. Dyskeratosis congenita with malignant transformation. BMJ Case Rep. 2011 Jan 11;2011:bcr0320102848. doi: 10.1136/bcr.03.2010.2848. PMID: 22715219; PMCID: PMC3029444
Ogden, G. R., Connor, E., & Chisholm, D. M. Dyskeratosis congenita: Report of a case and review of the literature. Oral Surgery, Oral Medicine, Oral Pathology, 1988; 65(5), 586–591. https://doi.org/10.1016/0030-4220(88)90142-9
McKay, G. S., Ogden, G. R., & Chisholm, D. M. (1991). Lingual hyperkeratosis in dyskeratosis congenita: Ultrastructural findings. Journal of Oral Pathology and Medicine, 20(4), 196–199. https://doi.org/10.1111/j.1600-0714.1991.tb00921.x
Ogden, G. R., Chisholm, D. M., Hopwood, D., & Lane, E. B. (1993). Evidence for field change in oral cancer based on cytokeratin expression. British Journal of Cancer, 67, 1324–1330. https://doi.org/10.1038/bjc.1993.245
Cannell H. Dyskeratosis congenita. Br J Oral Surg 1971;9: 8–10



