Nasljedni poremećaji koji nose povećan rizik za nastanak karcinoma usne šupljine zbog genetičke nestabilnosti su Fanconijeva anemija, Dyskeratosis congenita, Xeroderma pigmentosum, Li Fraumenijev sindrom, Bloomov sindrom, Ataxia telangiectasia i Cowdenov sindrom.
Fanconijeva anemija najsnažnije je povezana s predispozicijom za karcinom usne šupljine. Dyskeratosis congenita (DKC) (znana i kao Zinsser-Cole-Engmanov sindrom) je rijetki nasljedni poremećaj s predispozicijom za nastanak oralne leukoplakije koja se može maligno transformirati u ranoj životnoj dobi.
U ovom poglavlju opisat ćemo potencijalno zloćudne poreećaje oralne sluznice (PZPOS) i potencijal za zloćudnu preobrazbu u dva nasljedna stanja, Fanconijevoj anemiji i Dyskeratosis congenita. PZPOS povezani s ova dva sindroma su kronična oralna reakcija presatka protiv primatelja (cGVHD) (klasificirana kao PZPOS 2020. godine), oralni lihen planus i oralna leukoplakija.
Fanconijeva anemija
- Fanconijeva anemija (FA) je multisistemski poremećaj karakteriziran brojnim prirođenim abnormalnostima, progresivnim zatajenjem koštane srži i pancitopenijom te snažno izraženom sklonošću za razvoj akutne mijeloične leukemije (AML) i solidnih tumora.
- FA se javlja u svim dijelovima svijeta, od 1:100 000 poroda u SAD, 1:45 000 poroda u Izraelu do 1:22 000 poroda u Južnoj Africi. Incijencija je najviša među Romima u Španjolskoj.
- FA je klinički heterogen sindrom zatajenja koštane srži i prirođenih abnormalnosti koje mogu zahvatiti sve organske sustave
- Pacijenti s FA mogu imati čitav niz anomalija. Prirođene anomalije uključuju abnormalnosti skeleta (nizak rast, mikrocefalija, hipoplazija palca), abnormalnosti organa (bubrega, oka, uha, genitalija) i pigmentacije na koži poput bijele kave (café au lait pigmentacije) (Tablica 1).
- Do danas su identificirana 23 gena čija mutacija uzrokuje FA (FANCA-FANCV). Mutacije na genu FANCA su najčešće i javljaju se u 60-65% slučajeva. Sve mutacije na FA genima nasljeđuju se autosomno recesivno, osim mutacija na FANB i RAD51 genima koji se nasljeđuju preko X kromosoma, odnosno autosomno dominantno.
- FA protein sudjeluje u različitim aspektima popravka DNK, posebno u popravku križnih veza DNK, stabilizaciji genoma i regulaciji kaskadnih proteina. Poremećaj bilo kojeg FA proteina rezultira genomskom nestabilnošću, manjkavošću mehanizama popravka DNK i povećanim rizikom za nastanak raka.
- U pacijenata s FA i disfunkcijom koštane srži uobičajena metoda liječenja je transplantacija krvotvornih matičnih stanica (TKMS).
- Jedna od najčešćih komplikacija nakon TKMS je nastanak kronične reakcije presatka protiv primatelja (cGVHD) koja je također čimbenik rizika za nastanak karcinoma, posebno planocelularnog karcinoma. cGVHD je od 2020. godine klasificirana kao oralna potencijalno zloćudna lezija.
- Zbog velikog broja različitih kliničkih abnormalnosti, pacijenti s FFA zahtijevaju sveobuhvatnu dijagnostičku obradu.
- FA je ponekada teško razlikovati od drugih sindroma te može predstavljati izazov za dijagnozu. Šarolika klinička slika i varijabilna ekspresivnost u određenim slučajevima otežavaju ranu dijagnozu.
- Detaljan opis najčešćih poremećaja u pojedinim organima napravili su Moreno i sur. (2021).
- Laboratorijska dijagnoza bolesti postavlja testovima kromosomske lomljivosti.
- Ostale dijagnostičke metode uključuju western blot analizu, metodu istovremenog umnažanja vezanih proba i sekvencioniranje nove generacije.
- Nakon postavljanja dijagnoze potrebno je doživotno praćenje kako bi se što prije postavila dijagnoza karcinoma. Genetičko savjetovanje je od ključnog značaja.
KLINIČKI ZNAKOVI FANCONIJEVE ANEMIJE
(Nalepa i Clapp 2018)
- Kraniofacijalne malformacije
- Zaostajanje u razvoju
- Nizak rast
- Hipodoncija, mikrodoncija
- Krvarenje iz usne šupljine
- Otečene desni
- Abnormalnosti različitih organskih sustava
- Aplazija palca ili palčane kosti
- Osteoporoza
- Progresivno zatajenje koštane srži
- Srčane greške
- Malformacije urogenitalnog i gastrointenstinalnog sustava
- Displazija ili ektopija bubrega
- Disfunkcija endokrinih žlijezda
- Smanjena plodnost
- Četvrtini pacijenata koji razviju solidni tumor, FA se dijagnosticira nakon otkrića tumora.
- FA potrebno isključiti kod pojave tumora u mlađoj životnoj dobi. Opisano je nekoliko slučajeva oralnog karcinoma u mlađoj životnoj dobi u pacijenata s FA. Stoga bi u mladih pacijenata koji razviju karcinom glave i vrata a nemaju čimbenike rizika, trebalo razmotriti dijagnozu FA.
- FA može dovesti do zatajenja koštane srži, a transplantacija matičnih stanica može dovesti do cGVHD.
- Incidencija je 200-800 puta veća u pacijenata sa FA nego u općoj populaciji. U posljednje vrijeme, nastanak karcinoma usne šupljine u FA ponovo je pobudio pažnju istraživača.
- U pregledu literature, medijan dobi 12 pacijenata s FA koji su razvili karcinom nakon TKMS bio je 21 godinu, što je bilo značajno niže od 28 godina koliko su imali pacijenti s FA koji nisu bili liječeni TKMS (p
- Masserot i sur. (2008) opisali su 13 pacijenata s FA koji su razvili karcinomom glave i vrata nakon HSCT (od čega je 8 pacijenata razvilo karcinom usne šupljine), što je do sada najveća opisana skupina ovih pacijenata. Pojava karcinoma na jeziku u mlade osobe koja ne puši trebala bi pobuditi sumnju na FA kao podležeće medicinsko stanje i potaknuti na provođenje probira testovima lomljivosti kromosoma.
Dyskeratosis congenita
Nasljedna diskeratoza je rijetka multisistemska genodermatoza koja dovodi do smanjenja funkcije telomeraze narušavajući stabilnost kromosoma. Također poznata i kao Zinsser-Cole-Engman sindrom koji je prvi put opisan 1906. godine. Telomere na krajevima kromosoma i enzim telomeraza odgovorni su za sintezu telomera. Smanjena ili oštećena funkcija telomeraze dovodi do smanjenja genetskog materijala i naknadne aktivacije “puteva odgovora na oštećenje DNA” uzrokujući staničnu smrt.
Do zatajenja koštane srži dolazi jer je koštana srž ovisna o funkciji telomera zbog svoje visoke sposobnosti dijeljenja stanica. Osim toga, nasljedna diskeratoza se također često manifestira pojavom mukokutanih znakova te plućnom i/ili jetrenom fibrozom.
“Etiologija”
- Nasljedna diskeratoza jedan je od rijetkih nasljednih sindroma zatajenja koštane srži koja ima X-vezane, autosomno dominantne i autosomno recesivne obrasce nasljeđivanja.
- Specifični genetski defekt DKC1 mutacija udružena s X-vezanom nasljednom diskeratozom, najtežim oblikom nasljedne diskeratoze, dovodi do odsutnosti funkcioniranja diskerina, ozbiljno narušavajući funkciju telemeraze.
- Identificiran je niz genetskih mutacija koje su rezultirale različitim kliničkim manifestacijama, uključujući RTEL1 i TINF2 uz koje se kao istaknute značajke opisuje hipoplazija malog mozga i bolest mrežnice oka.
- Recesivne mutacije u NOP10, CTC1, NHP2, PARN i WRAP53 su rjeđe i dovode do različitih kliničkih slika.
“Epidemiologija”
- Nasljedna diskeratoza je rijetka, obično se klinički manifestira između 5-12 godine života i ima mušku predominaciju.
- Procjenjena incidencija iznosi 1 slučaj na 1000000 ljudi.
[/toggle]
“Klinička slika”
- Nasljedna diskeratoza se tipično javlja u ranoj životnoj dobi, za razliku od stečenih oblika aplastične anemije i posljedica je genetskih mutacija.
- Zinsser je 1906. godine prvi opisao karakterističnu trijadu nasljedne diskeratoze koja uključuje sljedeće:
- Retikularna atrofija i hiperpigmentacija kože
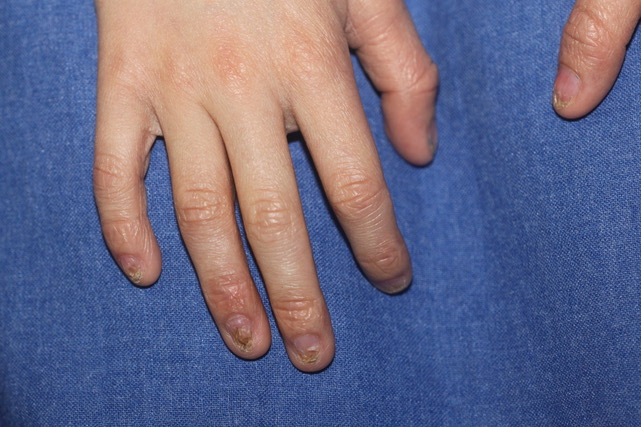
- Distrofija noktiju (Slika 1)
- Oralna leukoplakija
- U bolesnika s nasljednom diskeratozom, kao rezultat kromosomske nestabilnosti postoji povećan rizik od razvoja raka.
- Zbilježeno je da djeca s nasljednom diskeratozom imaju 100 puta veći rizik od razvoja akutne mijeloične leukemije.
- Također postoji povećan rizik od razvoja karcinoma pločastih stanica glave i vrata, grlića maternice i anogenitalne regije.
- Zabilježeni su i karcinomi kože, aerodigestivnog trakta i gušterače, a većina svih karcinoma javlja se u trećem desetljeću života.
[/toggle]
“Oralne Manifestacije”
- U bolesnika s nasljednom diskeratozom zabilježene su i promjene u usnoj šupljini uključujući oralnu leukoplakiju, koja je najčešća i javlja se u do 80% bolesnika.
- Najčešće zahvaćena mjesta su jezik, obrazna sluznica, nepce i gingiva.
- Također mogu nastati promjene mekih tkiva poput eritema, osobito na obraznoj sluznici i jeziku, te smeđa pigmentacija, i depapilacija jezika.
- Osim toga, zabilježena je i izmijenjena senzacija opisana kao žarenje jezika.
Dentalne implikacije:
- Upala gingive i parodontna bolest često su prisutne u bolesnika s nasljednom diskeratozom zbog kombinacije rane destrukcije parodontnog tkiva kao rezultat anomalija u strukturama ektodermalnog porijekla i slabog odgovora na to kao posljedica neutropenije.
- Karijes je često prisutan u bolesnika s nasljednom diskeratozom što potvrđuje i studija u kojoj je 13 od ukupno 73 bolesnika imalo opsežan karijes. Navedena činjenica potvrđena je i u drugim studijama.
- Ostale dentalne anomalije koje se viđaju u bolesnika s nasljednom diskeratozom uključuju hipodonciju, kratke zatupljene korijene, stanjenu caklinu, taurodontizam, smanjen omjer kruna/korijen, hipokalcifikaciju i recesiju gingive.
Zloćudna Probrazba:
- Zabilježena stopa zloćudne preobrazbe oralne leukoplakije kreće se između 0.13-34% ovisno o brojnim rizičnim čimbenicima poput dobi, spola, homogenosti i veličini lezije.
- Kod nasljedne diskeratoze stopa progresije takvih lezija u karcinom je približno 30% između 10-30 godina.
- Štoviše, kod nasljedne diskeratoze zbog ranog skraćivanja telomera, zabilježena je pojava karcinoma ranije u bolesnika mlađih od 5 godina. Stoga je pomno praćenje uz redovite biopsije neophodno.
[/toggle]
Diferencijalna dijagnoza
Aplastična anemija se može klasificirati u dvije široke kategorije; stečena i nasljedna. Stečeni uzroci uključuju idiopatske, što je najčešći uzrok, lijekom inducirane, virusne, trudničke bolesti i bolesti vezivnog tkiva. Nasljedni uzroci uključuju Fanconijevu anemiju, Shwachman-Diamond sindrom i GATA2 sindrom. Iako je Fanconijeva anemija i dalje najčešća diferencijalna dijagnoza.
Diferencijalne dijagnoze oralnih bijelih lezija uključuju infektivne procese kao što je kandidijaza i neinfektivne uzroke uključujući oralni lihen planus, lihenoidne lezije, bijeli spužvasti nevus, frikcijsku hiperkeratozu i reakciju presatka protiv primatelja (GVHD).
Dijagnoza
- Dijagnostički kriterija za nasljednu diskeratozu prema Dokal 2011 podrazumijeva prisutnost najmanje 2 od 4 glavne značajke (slabljenje funkcije koštane srži, abnormalna pigmentacija kože,distrofija nokta i leukoplakija) zajedno s najmanje još dvije identificirane somatske značajke. Iste su prikazane u tablici 1.
- Dijagnoza nasljedne diskeratoze može se potvrditi krvnim pretragama uključujući kompletnu krvnu sliku i broj retikulocita kako bi se utvrdilo je li prisutna pancitopenija.
- Biopsijom koštane srži može se detektirati hipocelularna srž.
- Daljnje istraživanja trebala bi uključiti specijalne testove poput genetskog sekvencioniranja, mjerenje duljine telomera protočnom citometrijom, fluorescentnu in situ hibridizaciju (FISH) i kompjutoriziranu tomografiju (CT) kako bi se utvrdilo jesu li zahvaćena pluća i/ili jetra.
- U slučaju pozitivne obiteljske anamneze prenatalna genetska dijagnostika uzorkovanjem korionskih resica ili genetska dijagnostika prije implantacije treba biti dostupna.
Liječenje
- Multidisciplinarni pristup je neophodan u njezi bolesnika s dijagnozom nasljedne diskeratoze, a zdravstveni djelatnici biti će uključeni prema zahvaćenosti organskih sustava.
- Hematolozi imaju glavnu ulogu budući je zatajenje koštane srži primarni uzrok ranog morbiditeta i smrtnosti.
- Oralna leukoplakija, osobito u bolesnika s nasljednom diskeratozom, može se transformirati u oralni karcinom te je stoga potrebno pozorno praćenje.
- Holistički pristup u prevenciji i liječenju dentalnih bolesti putem optimiziranih preventivnih savjeta i intervencija ključan je za poboljšanje kvalitete života bolesnika s nasljednom diskeratozom.
» Ne-kirurško Liječenje
Ne-Kirurško Liječenje
Ne-kirurško liječenje oralne leukoplakijetopikalno 0.1% otopinom tretinoina rezultiralo je djelomičnim poboljšanjem u dvoje srodnika u dobi od 10 i 15 godina. Kemoterapijski lijekovi poput bleomicina i ciklofosfamida pokazala su učinkovitost ali slični rezultati nisu zabilježeni u liječenju oralne leukoplakije. Drugi prijavljeni derivat vitamina A, etretinat , pokazao se učinkovitim za oralnu leukoplakiju kod nasljedne diskeratoze.
» View less
» Kirurško Liječenje
Kirurško Liječenje
Oralni i maksilofacijalni kirurzi mogu učiniti kirurški intervenirati ukoliko se otkrije atipija ili karcinom pločastih stanica. Resekcija mekih i tvrdih tkiva usne šupljine često će zahtijevati protetsku rekonstrukciju, stoga je detaljno planiranje s protetičarom neophodno kako bi se postigao zadovoljavajući funkcionalni i estetski rezultat.
| Klinički znakovi/abnormalnosti |
| Glavne/uobičajene karakteristike |
| Abnormalna pigmentacija kože |
| Distrofija nokta |
| Oslabljena funkcija koštane srži |
| Leukoplakija |
| Ostale prepoznate somatske značajke |
| Epifora |
| Poteškoće u učenju/kašnjenje u razvoju |
| Plućna bolest |
| Nizak rast |
| Ekstenzivni karijes/gubitak zuba |
| Suženje jednjaka |
| Prijevremeni gubitak kose/sijede/rijetke trepavice |
| Hiperhidroza |
| Tumori |
| Intrauterini zastoj u rastu |
| Jetrena bolest/peptički ulkus/enteropatija |
| Ataksija/hipoplazija malog mozga |
| Hipogonadizam/nespušteni testisi |
| Mikrocefalija |
| Striktura/fimoza uretre |
| Osteoporoza/aseptična nekroza/skolioza |
| Gluhoća |
Tablica 1: Kliničke značajke korištene u dijagnostičkim kriterijima nasljedne diskeratoze opisane prema Dokal (1).
» View less
Literatura
Ankathil R, Mathew A, Joseph F, Nair MK (1996). Is oral cancer susceptibility inherited? Report of five oral cancer families. Eur J Cancer B Oral Oncol. 32(1):63–7. https://doi.org/10.1016/0964-1955(95)00055-0 PMID:8729621
Garavello W, Foschi R, Talamini R, La Vecchia C, Rossi M, Dal Maso L, et al. (2008). Family history and the risk of oral and pharyngeal cancer. Int J Cancer. 122(8):1827–31. https://doi.org/10.1002/ijc.23199 PMID:18076043
Warnakulasuriya S, Kujan O, Aguirre-Urizar JM, Bagan JV, González-Moles MÁ, Kerr AR, Lodi G, Mello FW, Monteiro L, Ogden GR, Sloan P, Johnson NW. Oral potentially malignant disorders: A consensus report from an international seminar on nomenclature and classification, convened by the WHO Collaborating Centre for Oral Cancer. Oral Dis. 2020 Oct 31. doi: 10.1111/odi.13704. Epub ahead of print. PMID: 33128420.
Moreno OM, Paredes AC, Suarez-Obando F, Rojas A. An update on Fanconi anaemia: Clinical, cytogenetic and molecular approaches (Review). Biomed Rep. 2021 Sep;15(3):74. doi: 10.3892/br.2021.1450. Epub 2021 Jul 15. PMID: 34405046; PMCID: PMC8329995.
Nalepa G, Clapp DW. Fanconi anaemia and cancer: an intricate relationship. Nat Rev Cancer. 2018 Mar;18(3):168-185. doi: 10.1038/nrc.2017.116. Epub 2018 Jan 29. PMID: 29376519.
Alter BP. Cancer in Fanconi anemia, 1927-2001. Cancer. 2003 Jan 15;97(2):425-40. doi: 10.1002/cncr.11046. PMID: 12518367.
Masserot C, Peffault de Latour R, Rocha V, Leblanc T, Rigolet A, Pascal F, Janin A, Soulier J, Gluckman E, Socié G. Head and neck squamous cell carcinoma in 13 patients with Fanconi anemia after hematopoietic stem cell transplantation. Cancer. 2008 Dec 15;113(12):3315-22. doi: 10.1002/cncr.23954. PMID: 18831513.
Millen FJ, Rainey MG, Hows JM, Burton PA, Irvine GH, Swirsky D. Oral squamous cell carcinoma after allogeneic bone marrow transplantation for Fanconi anaemia. Br J Haematol. 1997 Nov;99(2):410-4. doi: 10.1046/j.1365-2141.1997.3683184.x. PMID: 9375763.
Furquim CP, Pivovar A, Amenábar JM, Bonfim C, Torres-Pereira CC. Oral cancer in Fanconi anemia: Review of 121 cases. Crit Rev Oncol Hematol. 2018 May;125:35-40. doi: 10.1016/j.critrevonc.2018.02.013. Epub 2018 Mar 6. PMID: 29650274.
Kutler DI, Auerbach AD, Satagopan J, Giampietro PF, Batish SD, Huvos AG, Goberdhan A, Shah JP, Singh B. High incidence of head and neck squamous cell carcinoma in patients with Fanconi anemia. Arch Otolaryngol Head Neck Surg. 2003 Jan;129(1):106-12. doi: 10.1001/archotol.129.1.106. PMID: 12525204.
Amenábar JM, Torres-Pereira CC, Tang KD, Punyadeera C. Two enemies, one fight: An update of oral cancer in patients with Fanconi anemia. Cancer. 2019 Nov 15;125(22):3936-3946. doi: 10.1002/cncr.32435. Epub 2019 Aug 7. PMID: 31390058.
Grein Cavalcanti L, Fuentes Araujo RL, Bonfim C, Torres-Pereira CC. Oral manifestations compatible with chronic graft-versus host disease in patients with Fanconi anemia. Biol Blood Marrow Transplant. 2015a;21:275-280
Grein Cavalcanti L, Lyko KF, Araújo RL, Amenábar JM, Bonfim C, Torres-Pereira CC. Oral leukoplakia in patients with Fanconi anaemia without hematopoietic stem cell transplantation. Pediatr Blood Cancer. 2015b Jun;62(6):1024-6. doi: 10.1002/pbc.25417. Epub 2015 Feb 14. PMID: 25682760.
Bongiorno, M., Rivard, S., Hammer, D., & Kentosh, J. Malignant transformation of oral leukoplakia in a patient with dyskeratosis congenita. Oral Surgery, Oral Medicine, Oral Pathology and Oral Radiology 2017; 124(4), e239–e24
Handley, T. P., & Ogden, G. R. Dyskeratosis congenita: Oral hyperkeratosis in association with lichenoid reaction. Journal of Oral Pathology and Medicine, 2006; 35(8), 508–512. https://doi.org/10.1111/j.1600-0714.2006.00434.x
Ray JG, Swain N, Ghosh R, Richa, Mohanty SP. Dyskeratosis congenita with malignant transformation. BMJ Case Rep. 2011 Jan 11;2011:bcr0320102848. doi: 10.1136/bcr.03.2010.2848. PMID: 22715219; PMCID: PMC3029444
Ogden, G. R., Connor, E., & Chisholm, D. M. Dyskeratosis congenita: Report of a case and review of the literature. Oral Surgery, Oral Medicine, Oral Pathology, 1988; 65(5), 586–591. https://doi.org/10.1016/0030-4220(88)90142-9
McKay, G. S., Ogden, G. R., & Chisholm, D. M. (1991). Lingual hyperkeratosis in dyskeratosis congenita: Ultrastructural findings. Journal of Oral Pathology and Medicine, 20(4), 196–199. https://doi.org/10.1111/j.1600-0714.1991.tb00921.x
Ogden, G. R., Chisholm, D. M., Hopwood, D., & Lane, E. B. (1993). Evidence for field change in oral cancer based on cytokeratin expression. British Journal of Cancer, 67, 1324–1330. https://doi.org/10.1038/bjc.1993.245
Cannell H. Dyskeratosis congenita. Br J Oral Surg 1971;9: 8–10



